Publication
- Home /
- Categories /
- Publication
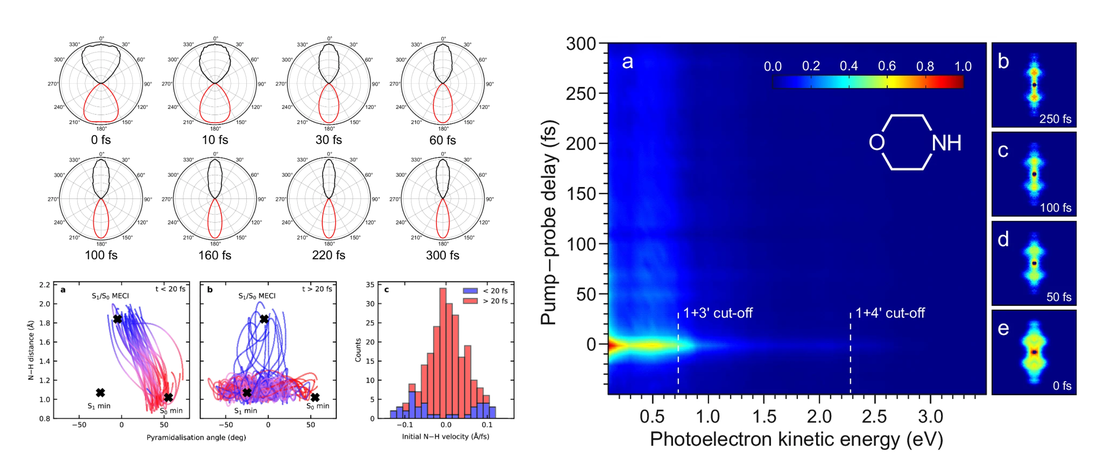
Decoupling structural molecular dynamics from excited state lifetimes using few-femtosecond ultraviolet resonant dispersive waves
- Sebastian L. Jackson, Andrew W. Prentice, Lauren Bertram, Lewis Hutton, Nikoleta Kotsina, Christian Brahms, Chris Sparling, John C. Travers, Adam Kirrander, Martin J. Paterson and Dave Townsend
- Publication
Optical sources exploiting resonant dispersive wave (RDW) emission are set to revolutionize ultrafast science. We demonstrate this approach by investigating excited state dynamics in morpholine using time-resolved photoelectron imaging…
Read More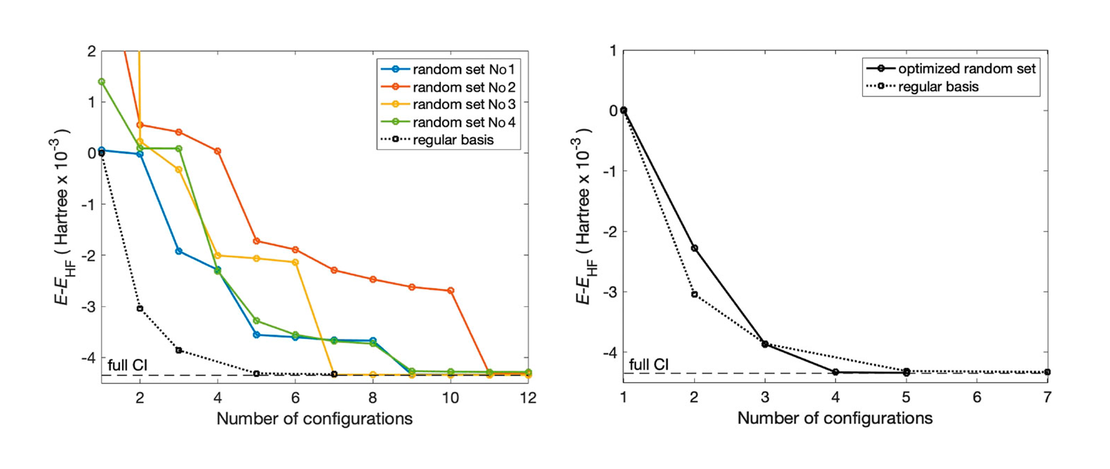
A new form of particle number conserving fermionic coherent states for electronic structure theory and electron dynamics
We propose a new form of Particle Number Conserving Fermionic Coherent States (PNCFCSs) that provide an efficient basis for calculating electronic wave functions. We demonstrate that a simple algorithm based on combinatorial analysis can be used for calculations of PNCFCS overlaps and matrix elements…
Read More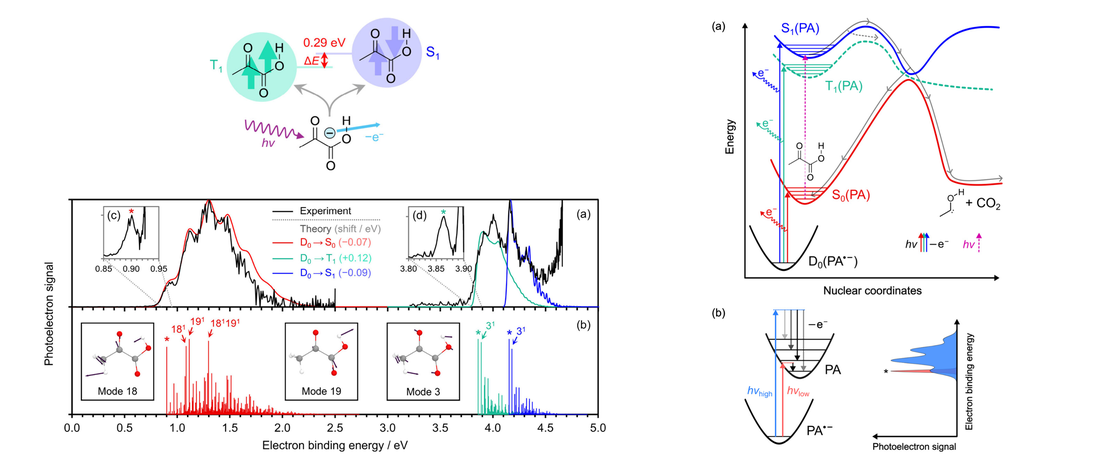
The Singlet–Triplet Gap of Pyruvic Acid
- E. Michi Burrow, Javier Carmona-García, Connor J. Clarke, Basile F. E. Curchod and Jan R. R. Verlet
- Publication
Understanding the gas-phase photochemistry of pyruvic acid is crucial to assessing its role and evolution in the atmosphere and relies on knowledge of the relative energy gaps between the atmospherically relevant excited electronic states of the molecule. However, accurate determination of these gaps, particularly between the lowest excited singlet and triplet states, has remained elusive…
Read More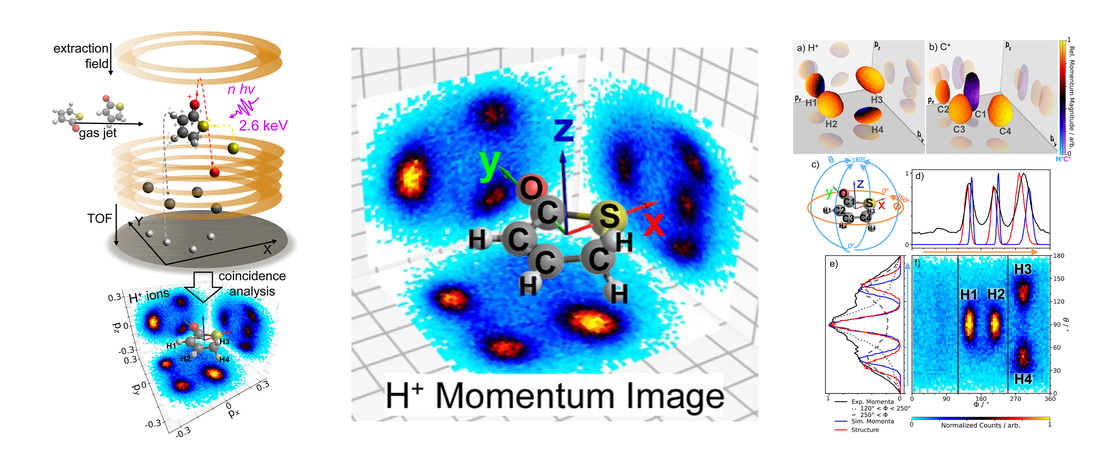
Visualizing the Three-Dimensional Arrangement of Hydrogen Atoms in Organic Molecules by Coulomb Explosion Imaging
- Alice E. Green, Keyu Chen, Surjendu Bhattacharyya, Felix Allum, Sergey Usenko, Michael N. R. Ashfold, Thomas M. Baumann, Kurtis D. Borne, Mark Brouard, Michael Burt, Basile F. E. Curchod, Benjamin Erk, Ruaridh J. G. Forbes, Lea M. Ibele, Rebecca A. Ingle, Huynh Van Sa Lam, Xiang Li, Kang Lin, Tommaso Mazza, Joseph W. McManus, Michael Meyer, Terence Mullins, Joao Pedro Figueira Nunes, Daniel E. Rivas, Aljoscha Roerig, Arnaud Rouzée, Philipp Schmidt, John Searles, Björn Senfftleben, Henrik Stapelfeldt, Rico Mayro P. Tanyag, Florian Trinter, Anbu Selvam Venkatachalam, Enliang Wang, Emily M. Warne, Peter M. Weber, Thomas J. A. Wolf, Till Jahnke, Artem Rudenko, Rebecca Boll and Daniel Rolles
- Publication , Project partner
Structure-sensitive methods based on femtosecond light or electron pulses are now making it possible to measure how molecular structures change during light-induced processes. Despite significant progress, high-fidelity imaging of nuclear positions remains a challenge even for relatively small molecular systems and, notably, regarding the positions of hydrogen atoms…
Read More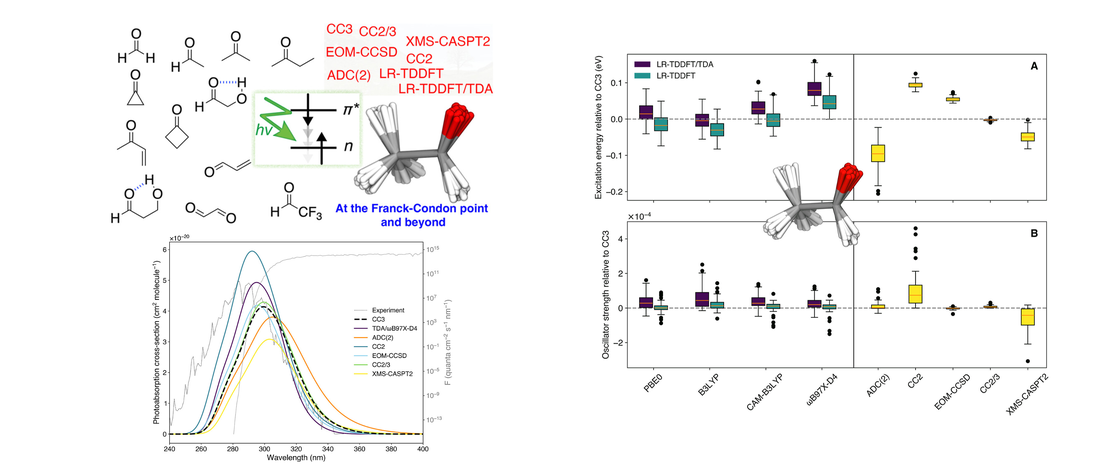
Benchmarking Electronic-Structure Methods for the Description of Dark Transitions in Carbonyls at and Beyond the Franck–Condon Point
Herein, we propose a comprehensive benchmark of electronic-structure methods to describe dark transitions, that is, transitions to excited electronic states characterized by a near-zero oscillator strength. This type of electronic state is particularly important for the photochemistry of molecules containing carbonyl groups, such as atmospheric volatile organic compounds (VOCs)…
Read More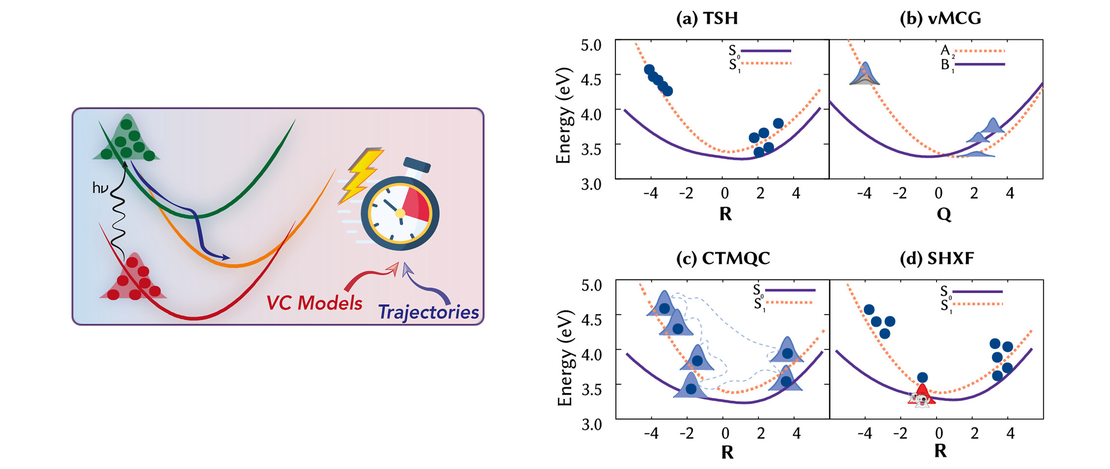
Perspective: Vibronic Coupling Potentials for Trajectory-Based Excited-State Dynamics
- Sandra Gómez, Patricia Vindel-Zandbergen, Dilara Farkhutdinova, and Leticia González
- Publication , Perspective , Project partner
This Perspective reviews the use of vibronic coupling (VC) potentials in trajectory-based excited-state dynamics simulations. Originally developed to provide simplified yet physically grounded representations of nonadiabatic interactions, VC models - particularly their linear version (LVC) - have facilitated extensive investigations of photophysical and photochemical processes, in both molecular and condensed-phase systems…
Read More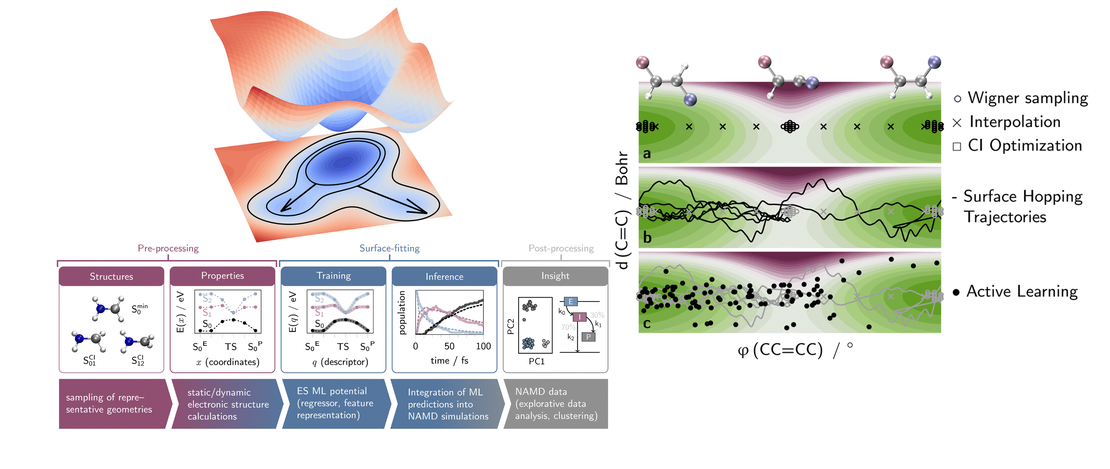
Machine learning for nonadiabatic molecular dynamics: best practices and recent progress
- Carolin Müller, Štěpán Sršeň, Brigitta Bachmair, Rachel Crespo-Otero, Jingbai Li, Sascha Mausenberger, Max Pinheiro, Jr, Graham Worth, Steven A. Lopez and Julia Westermayr
- Publication
Exploring molecular excited states holds immense significance across organic chemistry, chemical biology, and materials science. Understanding the photophysical properties of molecular chromophores is crucial for designing nature-inspired functional molecules, with applications ranging from photosynthesis to pharmaceuticals…
Read More
Is there anybody out there? Ultrafast Rydberg–valence interactions in the photodissociation of trimethylamine
- Derri J. Hughes, Andrew W. Prentice, Lauren Bertram, Richard T. Chapman, Luca Craciunescu, Daniel A. Horke, Peter Krüger, Michael A. Parkes, Henry J. Thompson, Emma Springate, James O. F. Thompson, Yu Zhang, Adam Kirrander, Martin J. Paterson, and Russell S. Minns
- Publication
Trimethylamine (TMA) is a tertiary aliphatic amine that stands as a potential marker for life beyond Earth due to only being naturally produced via biotic means. However, its propensity to undergo photodissociation in the gas phase when excited by a deep ultraviolet photon means that its amine daughter product could serve as an additional biomarker and confirmational spectral signature of TMA in exoplanetary atmospheres…
Read More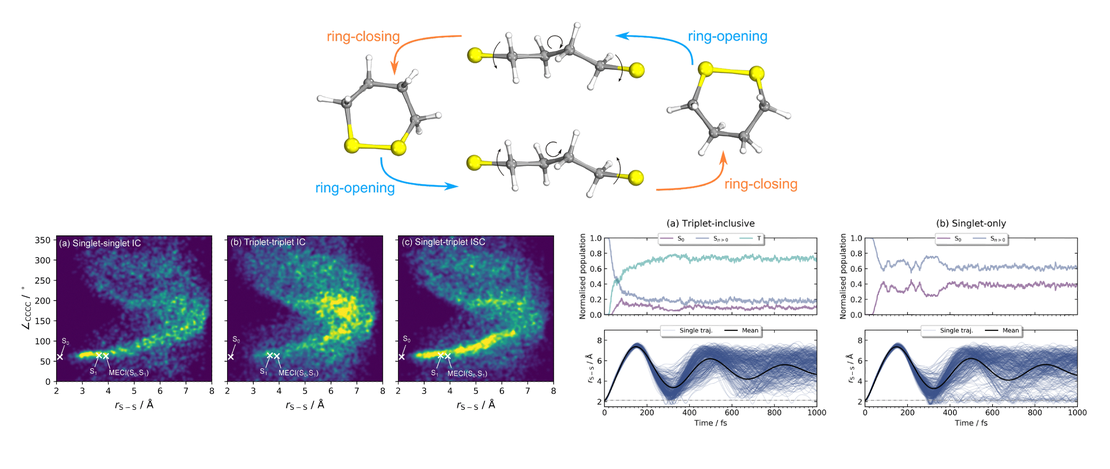
Triplets in the cradle: ultrafast dynamics in a cyclic disulfide
The effect of spin–orbit coupling on the ‘‘Newton’s cradle’’-type photodynamics in the cyclic disulfide 1,2-dithiane (C$ 4$H$ 8$S$ 2$) is investigated theoretically.We consider excitation by a 290 nm laser pulse and simulate the subsequent ultrafast nonadiabatic dynamics by propagating surface-hopping trajectories using SA(4|4)-CASSCF(6,4)-level electronic structure calculations with a modified ANO-R1 basis set…
Read More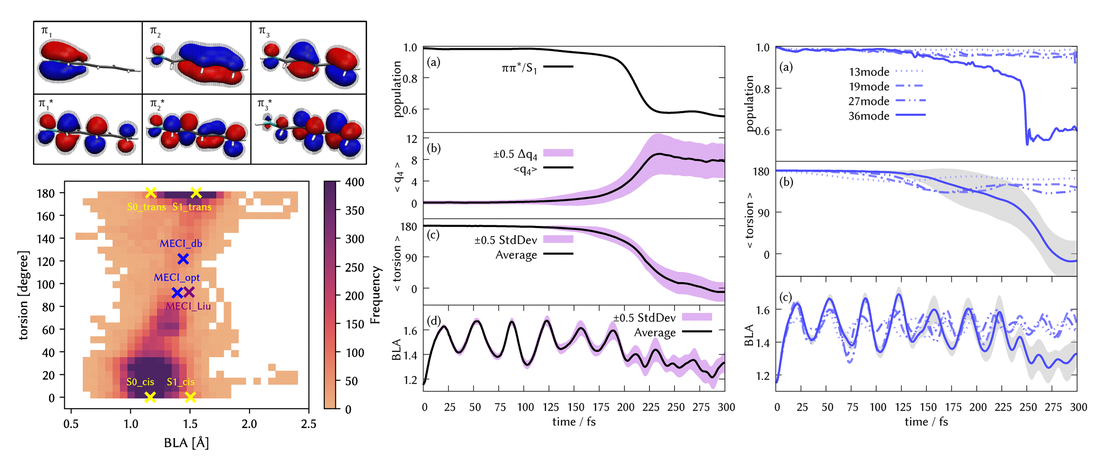
Delayed photoisomerisation of the trans-PSB3 retinal toy model using on-the-fly quantum dynamics
- María Mallo, Susana Gómez-Carrascoa and Sandra Gómez
- Publication , Project partner , Project collaboration
We explore the trans–cis photoisomerisation process in a representative retinal protonated Schiff base known as trans-PSB3, employing the quantum dynamics method direct dynamics variational multiconfigurational Gaussian -DD-vMCG- in full dimensionality, i.e., 36 degrees of freedom on potential energy surfaces computed on-the-fly using the SA(2)-CAS(6,6)SCF electronic structure method with the 6-31G basis set…
Read More
Tethered Gaussian wavepackets for quantum dynamics simulations: Sticking together for better convergence
Standard Gaussian products are frequently employed as a basis to represent potential energy surfaces (PESs) or time-dependent wavefunctions. The effective “volume” of standard Gaussian products decreases as dimensionality f increases, meaning that rapidly growing basis sets are required to obtain accurate results as f grows…
Read More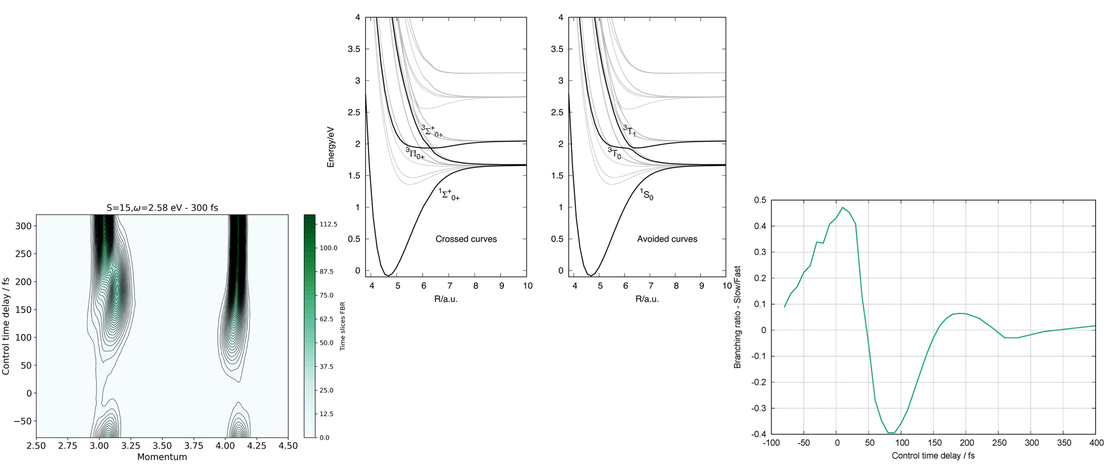
The strong-field control of IBr photodissociation re-visited
The photodissociation of IBr is a paradigm for a process that can be controlled by a strong, nonresonant electric field, known as the non-resonant dynamic stark effect (NRDSE). As shown by B. J. Sussman et al., Science, 2006, 314, 278, a carefully timed intense infra-red pulse can enhance or reduce the flux into the different dissociation channels…
Read More
Ultrafast ring-opening dynamics of 1,2-dithiane following ultraviolet absorption
- Patrick A. Robertson, James Merrick, David Heathcote, Matthew S. Robinson, Alexander Butler, Yasmine Biddick, J.F. Pedro Nunes, Conor Rankine, Zhihao Liu, Samuel F. Arrowsmith, James O.F. Thompson, M. Nrisimha Murty, Richard Chapman, Emma Springate, Edward A. Anderson, Adam Kirrander and Claire Vallance
- Publication
We report results from a recent laser pump–probe study into the ultrafast ring-opening dynamics of 1,2-dithiane. Following absorption of a 290 nm photon, the nuclear dynamics were probed as a function of pump–probe delay on the femtosecond timescale by strong-field ionisation with an 800 nm probe pulse, resulting in production of a range of atomic and molecular fragment ions…
Read More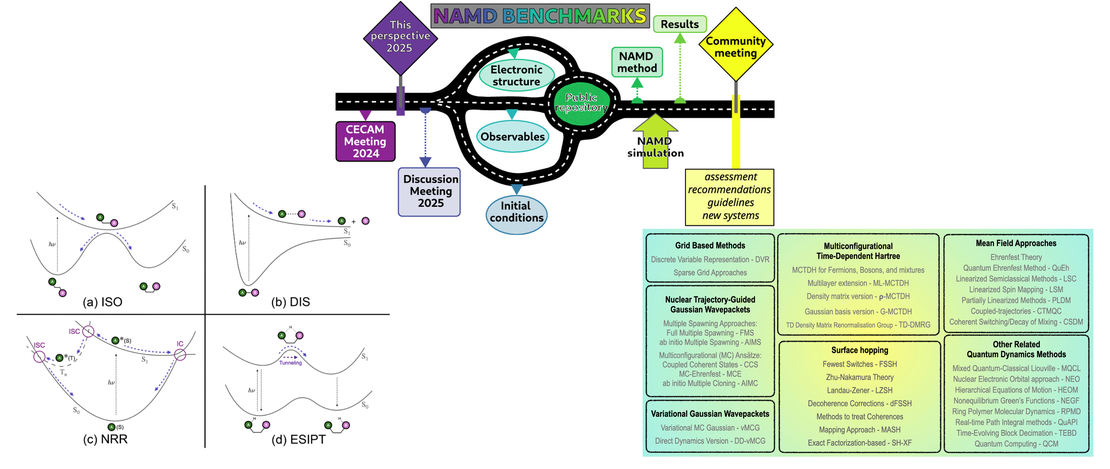
Roadmap for Molecular Benchmarks in Nonadiabatic Dynamics
- Léon L. E. Cigrang, Basile F. E. Curchod, Rebecca A. Ingle, Aaron Kelly, Jonathan R. Mannouch, Davide Accomasso, Alexander Alijah, Mario Barbatti, Wiem Chebbi, Nadja Došlić, Elliot C. Eklund, Sebastian Fernandez-Alberti, Antonia Freibert, Leticia González, Giovanni Granucci, Federico J. Hernández, Javier Hernández-Rodríguez, Amber Jain, Jiří Janoš, Ivan Kassal, Adam Kirrander, Zhenggang Lan, Henrik R. Larsson, David Lauvergnat, Brieuc Le Dé, Yeha Lee, Neepa T. Maitra, Seung Kyu Min, Daniel Peláez, David Picconi, Zixing Qiu, Umberto Raucci, Patrick Robertson, Eduarda Sangiogo Gil, Marin Sapunar, Peter Schürger, Patrick Sinnott, Sergei Tretiak, Arkin Tikku, Patricia Vindel-Zandbergen, Graham A. Worth, Federica Agostini, Sandra Gómez, Lea M. Ibele and Antonio Prlj
- Publication , Project collaboration , Perspective
Simulating the coupled electronic and nuclear response of a molecule to light excitation requires the application of nonadiabatic molecular dynamics. However, when faced with a specific photophysical or photochemical problem, selecting the most suitable theoretical approach from the wide array of available techniques is not a trivial task…
Read More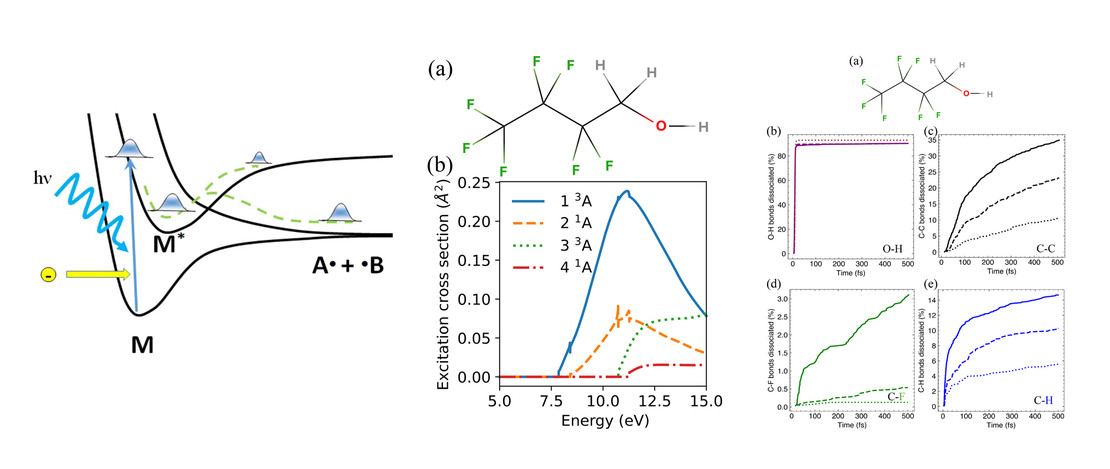
Rules of triplet state electron impact neutral dissociation in plasma from molecular dynamics simulations and an electrophore model
- Ryan Brook, Oliver Bramley, Dmitry V. Makhov, Anna Nelson, Gregory Armstrong, Joseph Yong, Ezri Saunders, Johnny de Viggiani, Jonathan Tennyson and Dmitrii V. Shalashilin
- Publication
Electron impact driven neutral dissociation of molecules that is important in low temperature plasma is investigated. Despite its importance for plasma technologies in microelectronics manufacturing, this process has received almost no attention from the computational chemistry community, which for decades has been focused on photodissociation…
Read More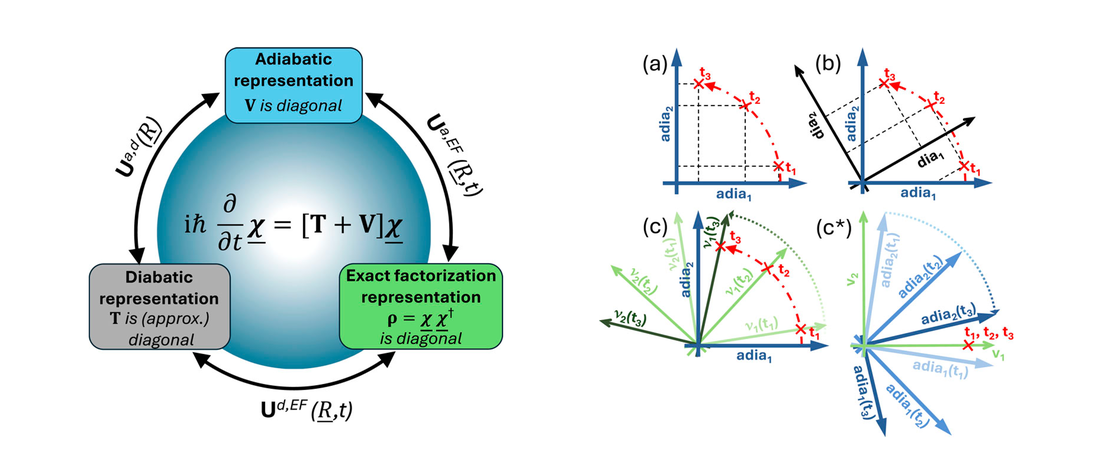
On the connection between the exact factorization and the Born–Huang representation of the molecular wavefunction
In this Note, we propose a new perspective on the foundations of the exact factorization (EF) representation of the molecular wavefunction,1,2 strengthening its connection to the more traditional Born–Huang (BH) representation, which invokes an expansion of the molecular wavefunction in terms of a basis of electronic states.3,4 In particular, we demonstrate that EF is the representation of the molecular wavefunction in terms of eigenstates of the density operator…
Read More
DONKEY: A Flexible and Accurate Algorithm for Clustering
We propose an accurate clustering algorithm suitable for the varied and multidimensional data sets that correspond to temporal snapshots from on-the-fly nonadiabatic trajectory-based simulations of photoexcited dynamics. The algorithm approximates the underlying probability density function using variable kernel density estimation, with local maxima corresponding to cluster centers…
Read More
Capturing Ultrafast Spin Dynamics in Single-Molecule Magnets Using Femtosecond X‑ray Emission Spectroscopy
- Kyle Barlow, Ryan Phelps, Julien Eng, Rebecca A. Ingle, Dmitry Khakhulin, Mykola Biednov, Sharmistha Paul Dutta, Yifeng Jiang, Frederico A. Lima, Vandana Tiwari, Christopher Milne, Tetsuo Katayama, Marco Coletta, Euan K. Brechin, Thomas J. Penfold and J. Olof Johansson
- Publication , Project collaboration
Achieving ultrafast photomagnetic switching of single-molecule magnets (SMMs) could lead to simultaneous fast and dense data storage devices. To facilitate this, a thorough understanding of the ultrafast dynamics emerging after ultrashort laser pulse excitation is essential. However, the complex nature of these materials means there is a lack of established experimental techniques that can probe the spin dynamics in SMMs…
Read More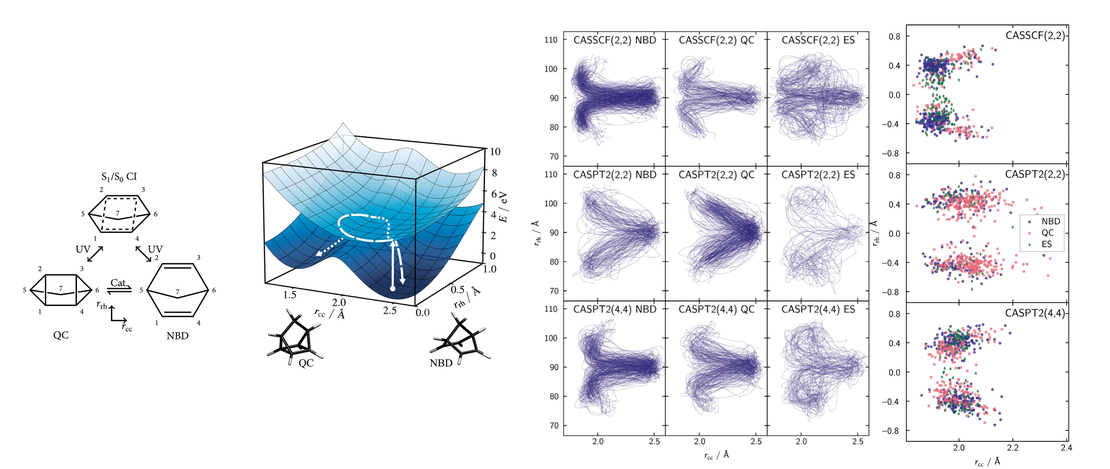
Photoexcited dynamics of the valence states of norbornadiene
The non-radiative decay of photoexcited norbornadiene, which together with its isomer quadricyclane forms a molecular photoswitch, is investigated using surface-hopping non-adiabatic dynamics. The simulations are performed using four levels of electronic structure theory: CASSCF(2,2), CASSCF(4,4), XMS-CASPT2(2,2), and XMS-CASPT2(4,4)…
Read More
Selecting Initial Conditions for Trajectory-Based Nonadiabatic Simulations
In this Account, we aim to warn the experienced or potential user of nonadiabatic molecular dynamics about the possible limitations of this strategy for initial-condition generation and its inability to accurately describe the photoexcitation of a molecule. More specifically, we argue that the initial phase-space distribution can be more accurately represented through molecular dynamics simulations by using a quantum thermostat.
Read More
Revealing the reaction path of UVC bond rupture in cyclic disulfides with ultrafast x-ray scattering
- Lingyu Ma, Wenpeng Du, Haiwang Yong, Brian Stankus, Jennifer M. Ruddock, Andrés Moreno Carrascosa, Nathan Goff, Yu Chang, Nikola Zotev, Darren Bellshaw, Thomas J. Lane, Mengning Liang, Sébastien Boutet, Sergio Carbajo, Joseph S. Robinson, Jason E. Koglin, Michael P. Minitti, Adam Kirrander, Theis I. Sølling, and Peter M. Weber
- Publication
Disulfide bonds are ubiquitous molecular motifs that influence the tertiary structure and biological functions of many proteins. Yet, it is well known that the disulfide bond is photolabile when exposed to ultraviolet C (UVC) radiation…
Read More
Assessing Nonadiabatic Dynamics Methods in Long Timescales
- Saikat Mukherjee, Yorick Lassmann, Rafael S. Mattos, Baptiste Demoulin, Basile F. E. Curchod and Mario Barbatti
- Publication
Nonadiabatic dynamics simulations complement time-resolved experiments by revealing ultrafast excited-state mechanistic information in photochemical reactions. Understanding the relaxation mechanisms of photoexcited molecules finds application in energy, material, and medicinal research…
Read More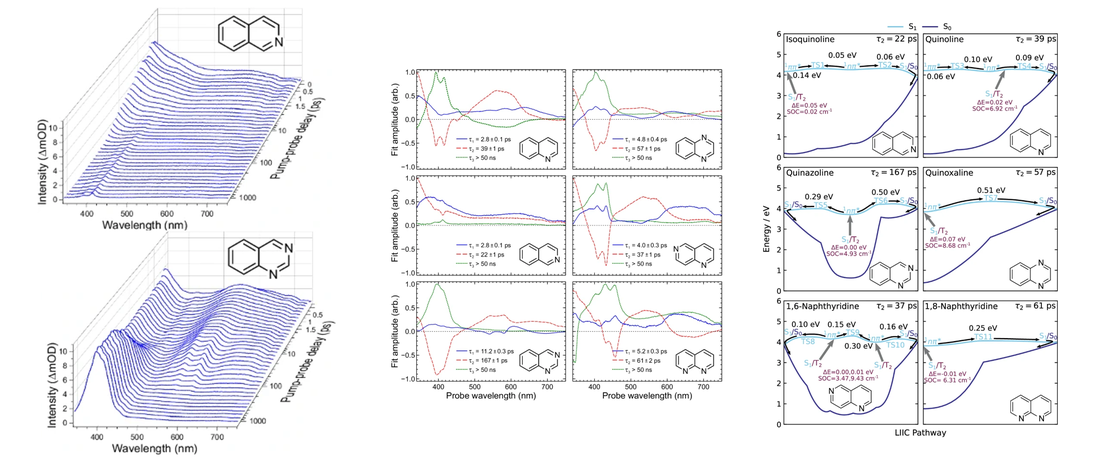
Excited state dynamics of azanaphthalenes reveal opportunities for the rational design of photoactive molecules
- Malcolm Garrow, Lauren Bertram, Abi Winter, Andrew W. Prentice, Stuart W. Crane, Paul D. Lane, Stuart J. Greaves, Martin J. Paterson, Adam Kirrander and Dave Townsend
- Publication
Various photoactive molecules contain motifs built on aza-aromatic heterocycles, although a detailed understanding of the excited state photophysics and photochemistry in such systems is not fully developed. To help address this issue, the non-adiabatic dynamics operating in azanaphthalenes under hexane solvation was studied following 267 nm excitation using ultrafast transient absorption spectroscopy…
Read More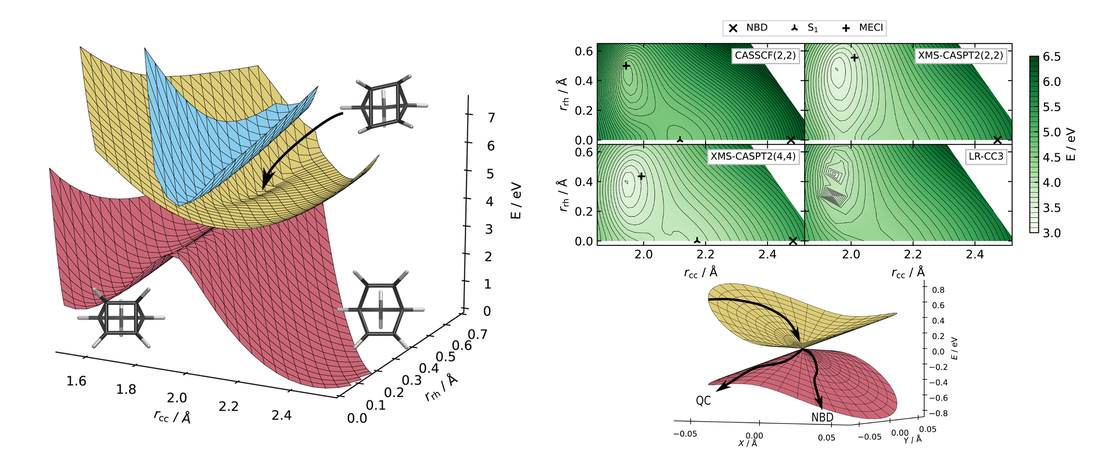
Electronic structure of norbornadiene and quadricyclane
The ground and excited state electronic structure of the molecular photoswitches quadricyclane and norbornadiene is examined qualitatively and quantitatively. A new custom basis set is introduced, optimised for efficient yet accurate calculations…
Read More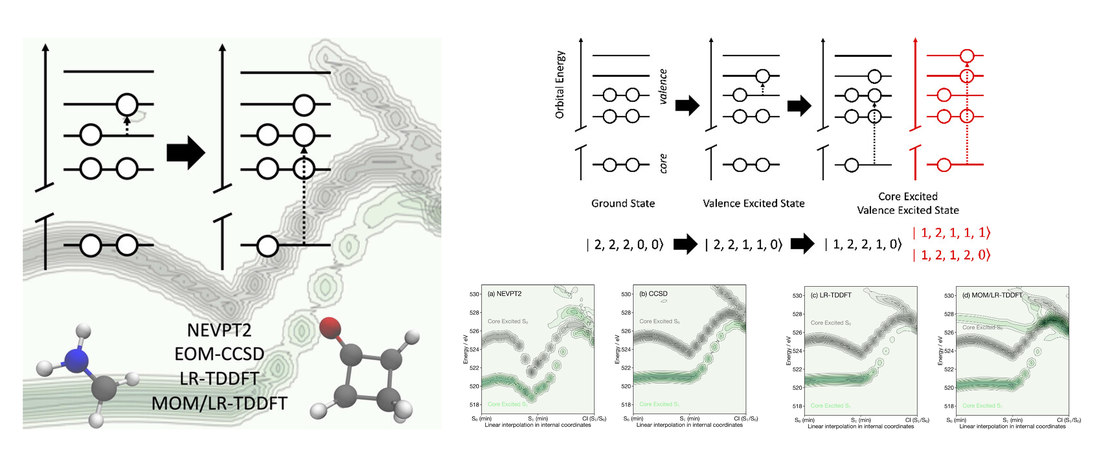
Exploring the Influence of Approximations for Simulating Valence Excited X‑ray Spectra
First-principles simulations of excited-state X-ray spectra are becoming increasingly important to interpret the wealth of electronic and geometric information contained within femtosecond X-ray absorption spectra recorded at X-ray Free Electron Lasers (X-FELs). However, because the transition dipole matrix elements must be calculated between two excited states (i.e., the valence excited state and the final core excited state arising from the initial valence excited state) of very different energies, this can be challenging and time-consuming to compute…
Read More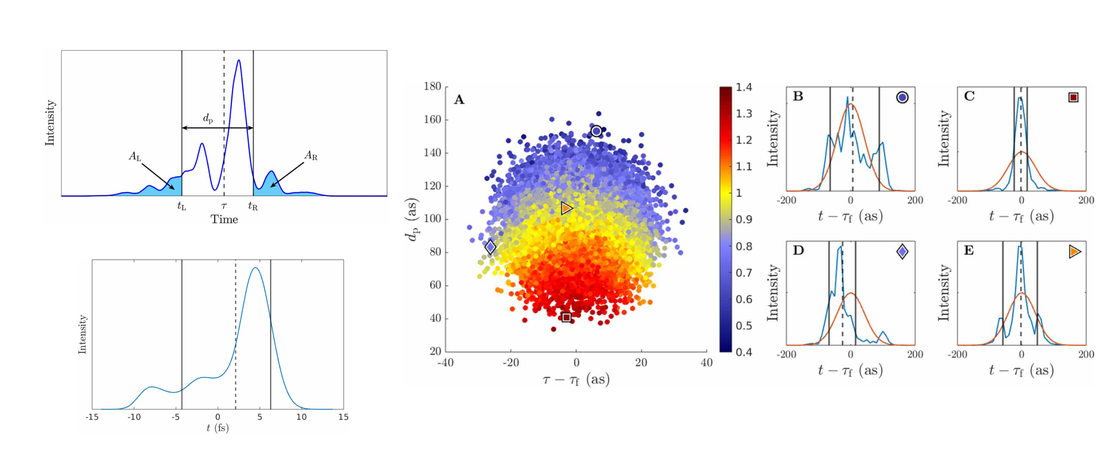
XFEL SASE pulses can enhance time-dependent observables
X-ray free electron lasers (XFELs) have emerged as powerful sources of short and intense x-ray pulses. We propose a simple and robust procedure which takes advantage of the inherent stochasticity of self-amplified stimulated emission (SASE) pulses to enhance the time-resolution and signal strength of the recorded data…
Read More
Rigid and planar π-conjugated molecules leading to long-lived intramolecular charge-transfer states exhibiting thermally activated delayed fluorescence
- Suman Kuila, Hector Miranda-Salinas, Julien Eng, Chunyong Li, Martin R. Bryce, Thomas J. Penfold and Andrew P. Monkman
- Publication
Intramolecular charge transfer (ICT) is a fundamental chemical process whereby excitation moves charge from an electron donor to an electron acceptor within the same molecule. Thermally activated delayed fluorescence (TADF) exploits the ICT property to harvest triplet excited states, leading to extensive optoelectronic applications, including OLEDs…
Read More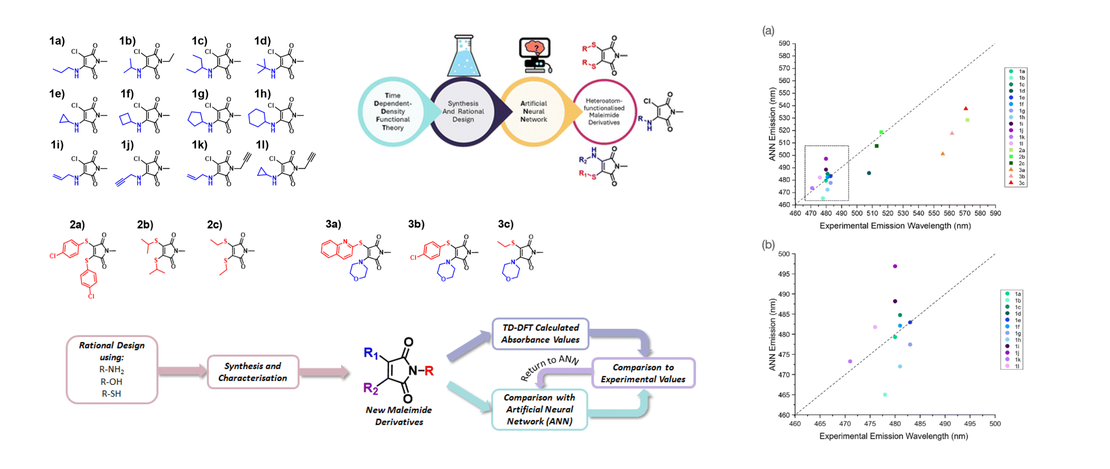
Integrated computational and experimental design of fluorescent heteroatom-functionalised maleimide derivatives
- Jake E. Barker, Gareth W. Richings, Yujie Xie, Julia Y. Rho, Calum T. J. Ferguson, Rachel K. O'Reilly and Scott Habershon
- Publication
The bottom-up design and synthesis of organic molecular species with tailored photophysical properties stands as an important challenge to both computational and experimental chemical science. Overcoming this challenge presents the potential to usher in new tools and approaches to improve our ability to develop new technologies, such as molecular sensors and attuned molecular switches…
Read More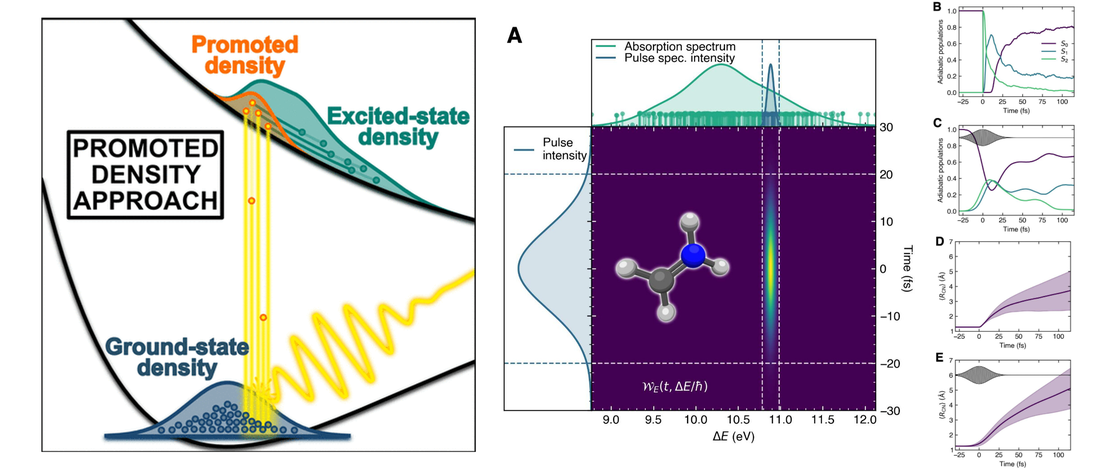
Including Photoexcitation Explicitly in Trajectory-Based Nonadiabatic Dynamics at No Cost
Over the last decades, theoretical photochemistry has produced multiple techniques to simulate the nonadiabatic dynamics of molecules. Surprisingly, much less effort has been devoted to adequately describing the first step of a photochemical or photophysical process: photoexcitation…
Read More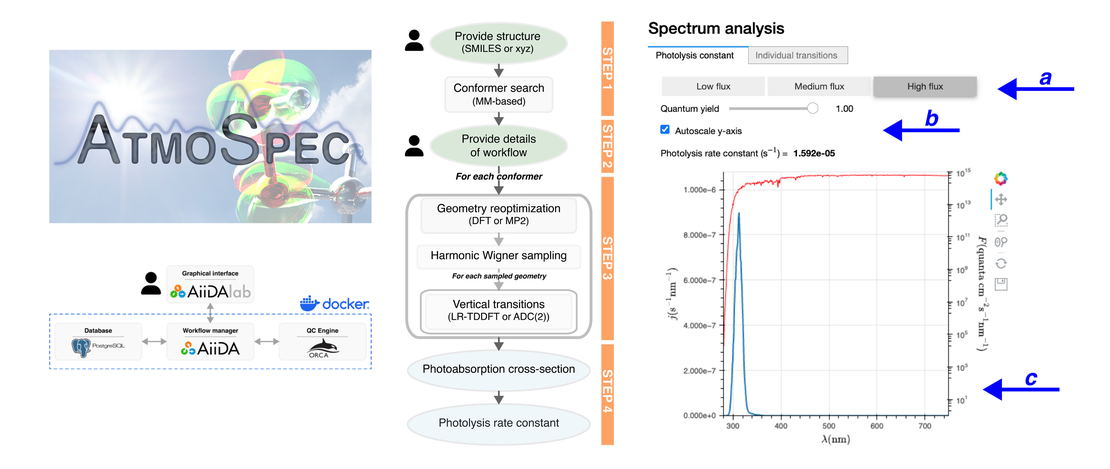
ATMOSPEC−A Tool to Calculate Photoabsorption Cross-Sections for Atmospheric Volatile Organic Compounds
Characterizing the photolysis processes undergone by transient volatile organic compounds (VOCs) in the troposphere requires the knowledge of their photoabsorption crosssection−quantities often challenging to determine experimentally, particularly due to the reactivity of these molecules…
Read More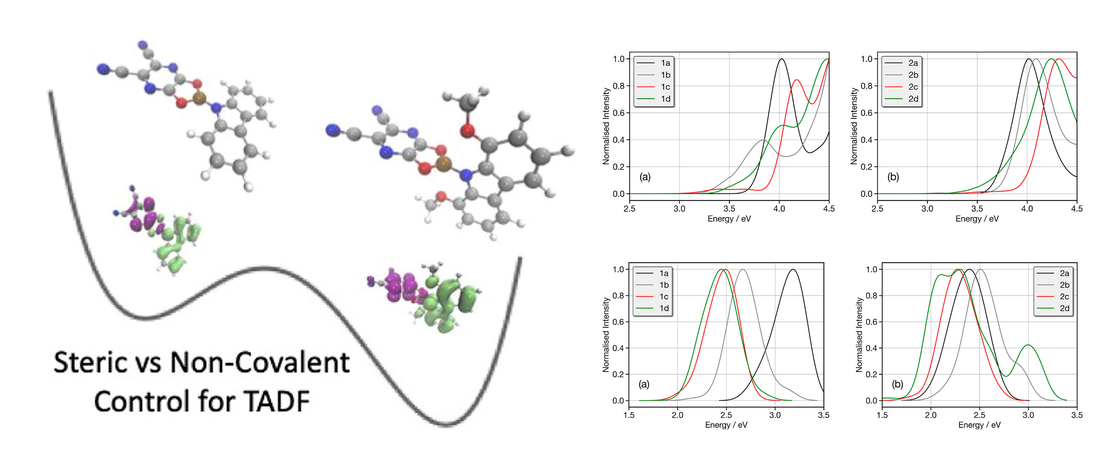
Conformational Control of Donor–Acceptor Molecules Using Non-covalent Interactions
Controlling the architecture of organic molecules is an important aspect in tuning the functional properties of components in organic electronics. For purely organic thermally activated delayed fluorescence (TADF) molecules, design is focused upon orthogonality orientated donor and acceptor units…
Read More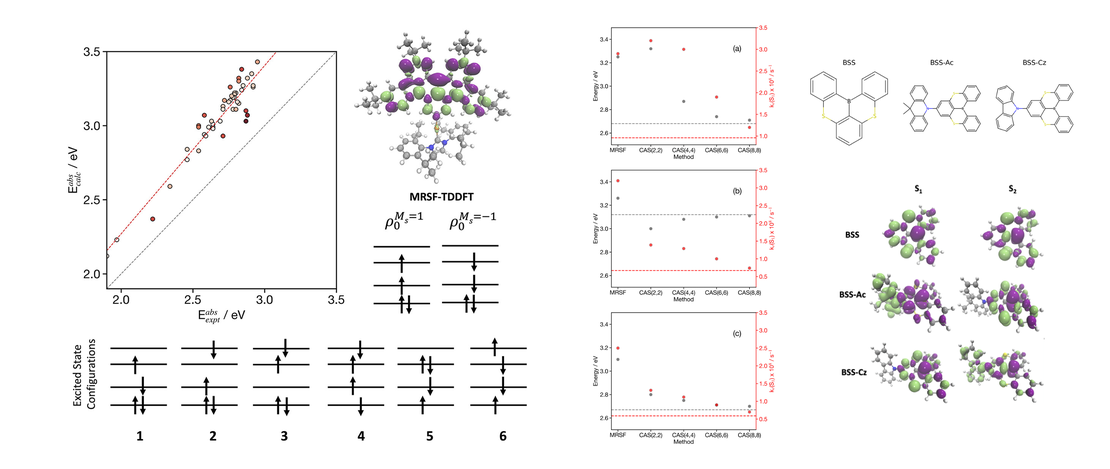
Towards the accurate simulation of multi-resonance emitters using mixed-reference spin-flip time-dependent density functional theory
Multi-resonant Thermally Activated Delayed Fluorescent (MR-TADF) materials have received significant research interest owing to their potential use as emitters in high-performance Organic Light Emitting Diodes (OLEDs). Despite their advantages, including narrow emission spectra leading to high colour purity, several challenges remain in optimising the performance of these materials…
Read More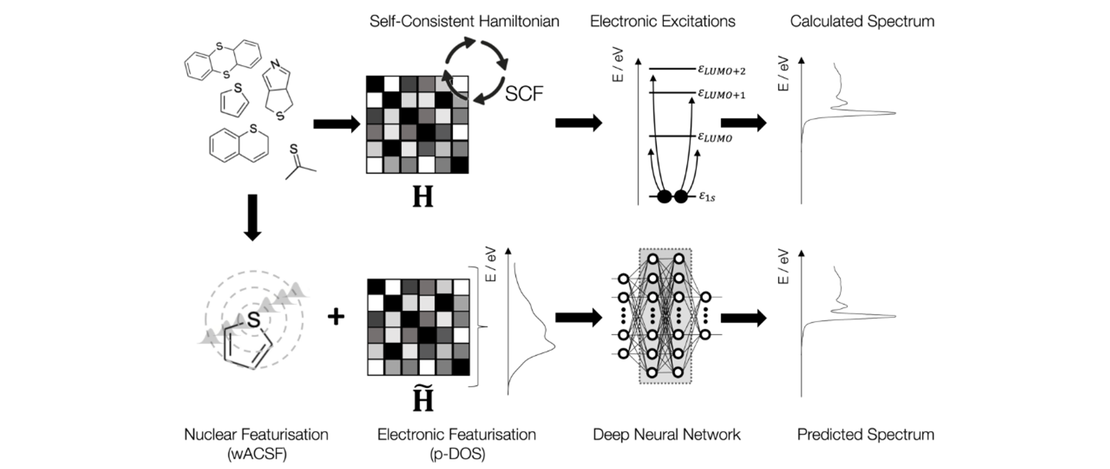
Partial Density of States Representation for Accurate Deep Neural Network Predictions of X-ray Spectra
The performance of a Machine Learning (ML) algorithm for chemistry is highly contingent upon the architect’s choice of input representation. This work introduces the partial density of states (p-DOS) descriptor: a novel, quantuminspired structural representation which encodes relevant electronic information for machine learning models seeking to simulate X-ray spectroscopy…
Read More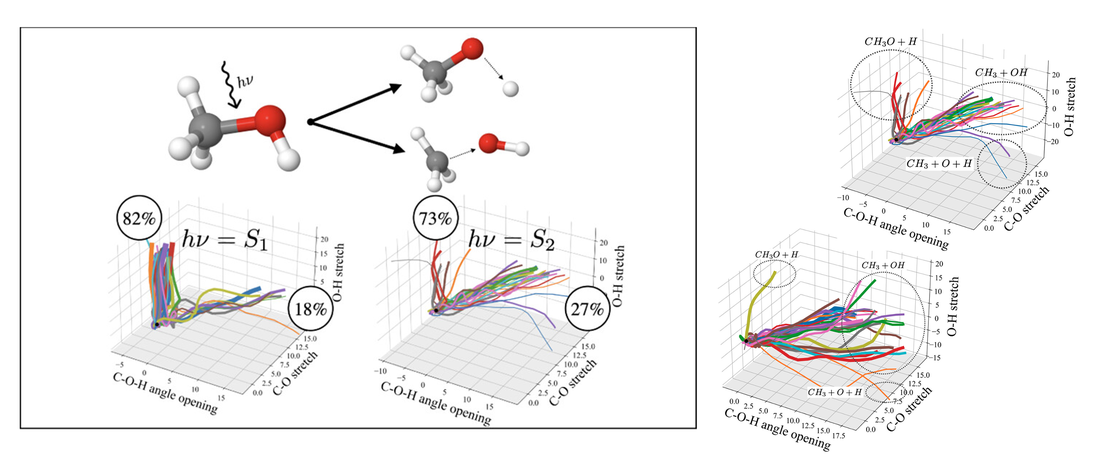
Modeling Photodissociation: Quantum Dynamics Simulations of Methanol
A comprehensive computational study of the gas-phase photodissociation dynamics of methanol is presented. Using a multiconfigurational active space based method (RASSCF) to obtain multidimensional potential energy surfaces (PESs) on-the-fly, direct quantum dynamics simulations were run using the variational multi-configurational Gaussian method (DD-vMCG)…
Read More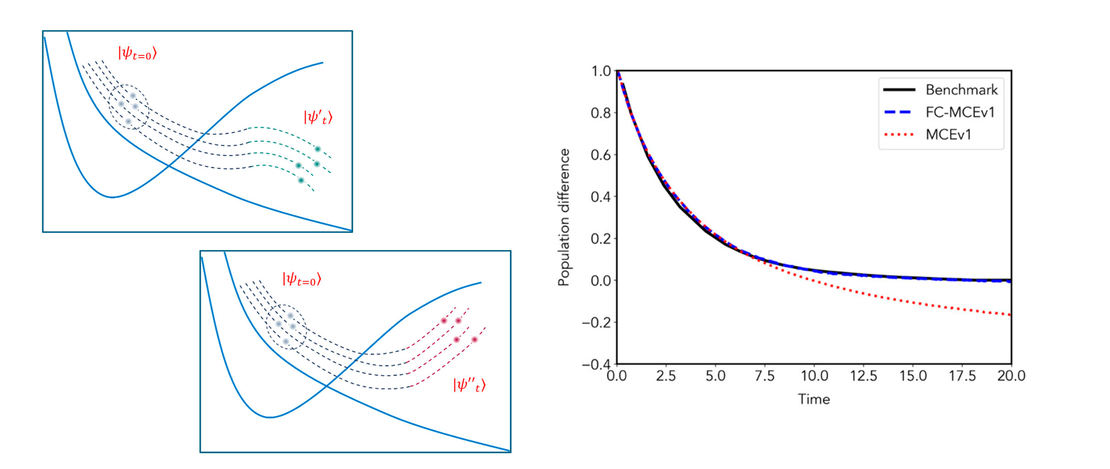
Full wave function cloning for improving convergence of the multiconfigurational Ehrenfest method: Tests in the zero-temperature spin-boson model regime
In this paper, we report a new algorithm for creating an adaptive basis set in the Multiconfigurational Ehrenfest (MCE) method, which is termed Full Cloning (FC), and test it together with the existing Multiple Cloning (MC) using the spin-boson model at zero-temperature as a benchmark…
Read More
Perspective on Theoretical and Experimental Advances in Atmospheric Photochemistry
Research that explores the chemistry of Earth’s atmosphere is central to the current understanding of global challenges such as climate change, stratospheric ozone depletion, and poor air quality in urban areas. This research is a synergistic combination of three established domains: earth observation, for example, using satellites, and in situ field measurements; computer modeling of the atmosphere and its chemistry; and laboratory measurements of the properties and reactivity of gas-phase molecules and aerosol particles…
Read More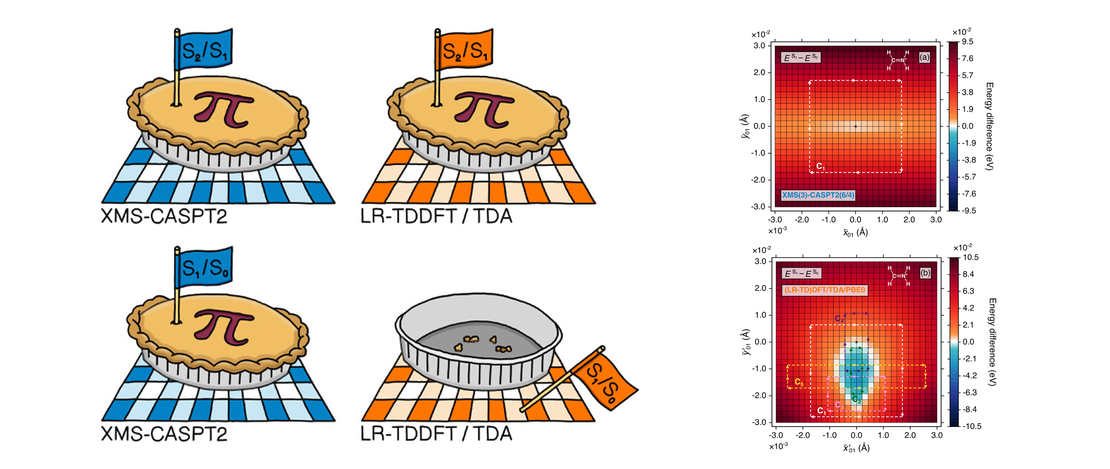
On the Topological Phase around Conical Intersections with Tamm–Dancoff Linear-Response Time-Dependent Density Functional Theory
Regions of nuclear-configuration space away from the Franck–Condon geometry can prove problematic for some electronic structure methods, given the propensity of such regions to possess conical intersections, i.e., (highly connected) points of degeneracy between potential energy surfaces. With the likelihood (perhaps even inevitability) for nonadiabatic dynamics simulations to explore molecular geometries in close proximity to conical intersections, it is vital that the performance of electronic structure methods is routinely examined in this context…
Read More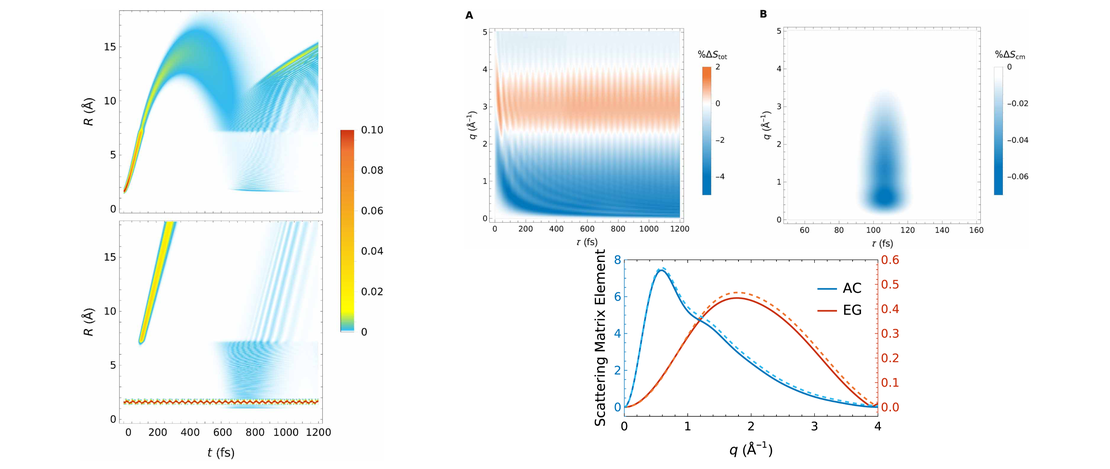
Ultrafast x-ray scattering and electronic coherence at avoided crossings: complete isotropic signals
Nonadiabatic transitions at conical intersections and avoided crossings play a pivotal role in shaping the outcomes of photochemical reactions. Using the photodissociation of LiF as a model, this theoretical study explores the application of gas phase nonresonant ultrafast x-ray scattering to map nonadiabatic transitions at an avoided crossing, utilizing the part of the scattering signal that probes electronic coherence directly…
Read More
Machine-learning strategies for the accurate and efficient analysis of x-ray spectroscopy
- Thomas Penfold, Luke Watson, Clelia Middleton, Tudur David, Sneha Verma, Thomas Pope, Julia Kaczmarek and Conor Rankine
- Publication
Computational spectroscopy has emerged as a critical tool for researchers looking to achieve both qualitative and quantitative interpretations of experimental spectra. Over the past decade, increased interactions between experiment and theory have created a positive feedback loop that has stimulated developments in both domains…
Read More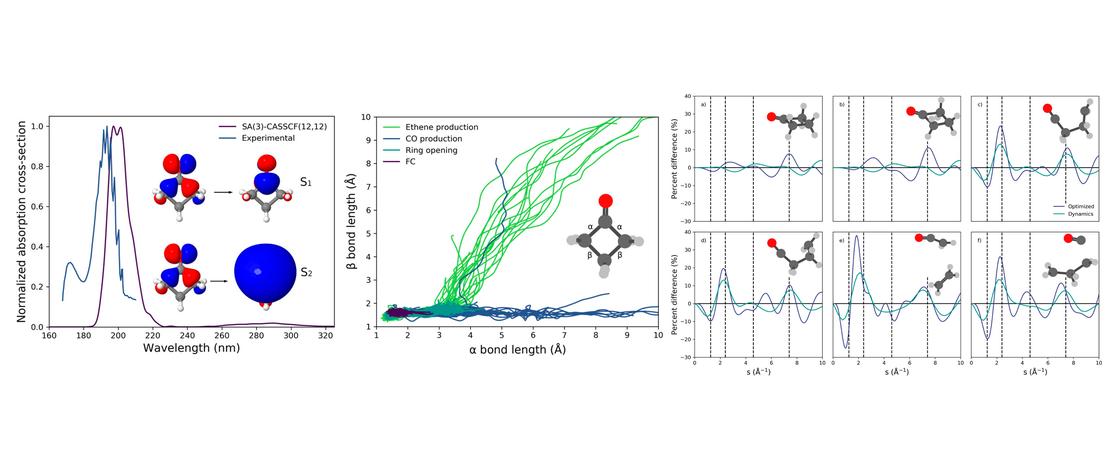
Using a multistate mapping approach to surface hopping to predict the ultrafast electron diffraction signal of gas-phase cyclobutanone
- Lewis Hutton, Andrés Moreno Carrascosa, Andrew W. Prentice, Mats Simmermacher, Johan E. Runeson, Martin J. Paterson and Adam Kirrander
- Publication , Prediction challenge
Using the recently developed multistate mapping approach to surface hopping (multistate MASH) method combined with SA(3)- CASSCF(12,12)/aug-cc-pVDZ electronic structure calculations, the gas-phase isotropic ultrafast electron diffraction (UED) of cyclobutanone is predicted and analyzed…
Read More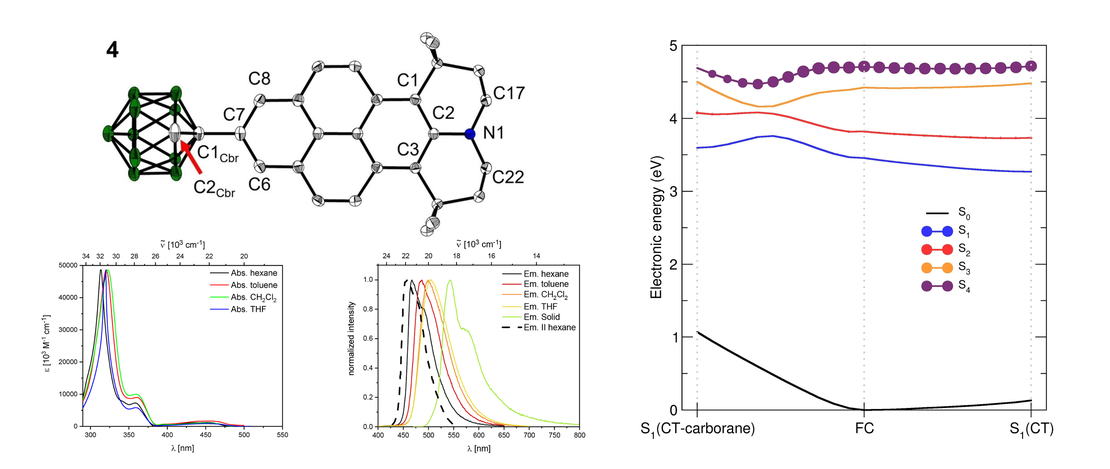
Synthesis, Photophysical and Electronic Properties of a D-π-A Julolidine-Like Pyrenyl-o-Carborane
- Johannes Krebs, Lisa Brändler, Ivo Krummenacher, Alexandra Friedrich, Holger Braunschweig, Maik Finze, Basile F. E. Curchod and Todd B. Marder
- Publication
We synthesized 2-(1-1,2-dicarbadodecaboranyl(12))-6,6,12,12-tetramethyl-7,8,11,12-tetrahydro-6H,10H-phenaleno[1,9-fg]pyrido[3,2,1-ij]quinoline (4), a julolidine-like pyrenyl-o-carborane, with pyrene substituted at the 2,7-positions on the HOMO/LUMO nodal plane. Using solid state molecular structures, photophysical data, cyclic voltammetry, DFT and LR-TDDFT calculations, we compare o-carborane and B(Mes)2 (Mes=2,4,6-Me3C6H2) as acceptor groups…
Read MoreTracking nuclear motion in single-molecule magnets using femtosecond X-ray absorption spectroscopy
- Kyle Barlow, Ryan Phelps, Julien Eng, Tetsuo Katayama, Erica Sutcliffe, Marco Coletta, Euan K. Brechin, Thomas J. Penfold and J. Olof Johansson
- Publication
The development of new data storage solutions is crucial for emerging digital technologies. Recently, all-optical magnetic switching has been achieved in dielectrics, proving to be faster than traditional methods. Despite this, single-molecule magnets (SMMs), which are an important class of magnetic materials due to their nanometre size, remain underexplored for ultrafast photomagnetic switching…
Read More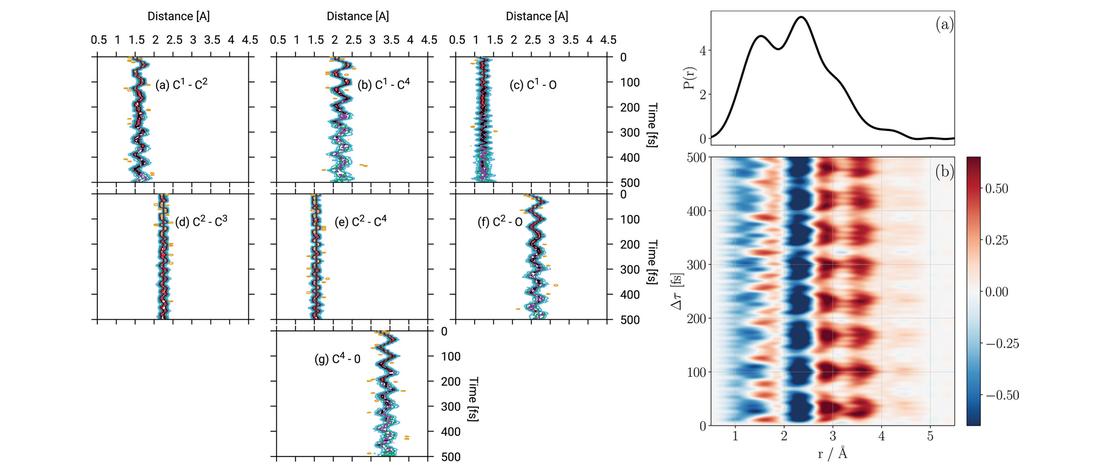
Prediction Through Quantum Dynamics Simulations: Photo-excited Cyclobutanone
- Olivia Bennett, Antonia Freibert, K. Eryn Spinlove and Graham A. Worth
- Prediction challenge , Publication
Quantum dynamics simulations are becoming a standard tool for simulating photo-excited molecular systems involving a manifold of coupled states, known as non-adiabatic dynamics. While these simulations have had many successes in explaining experiments and giving details of non-adiabatic transitions, the question remains as to their predictive power…
Read More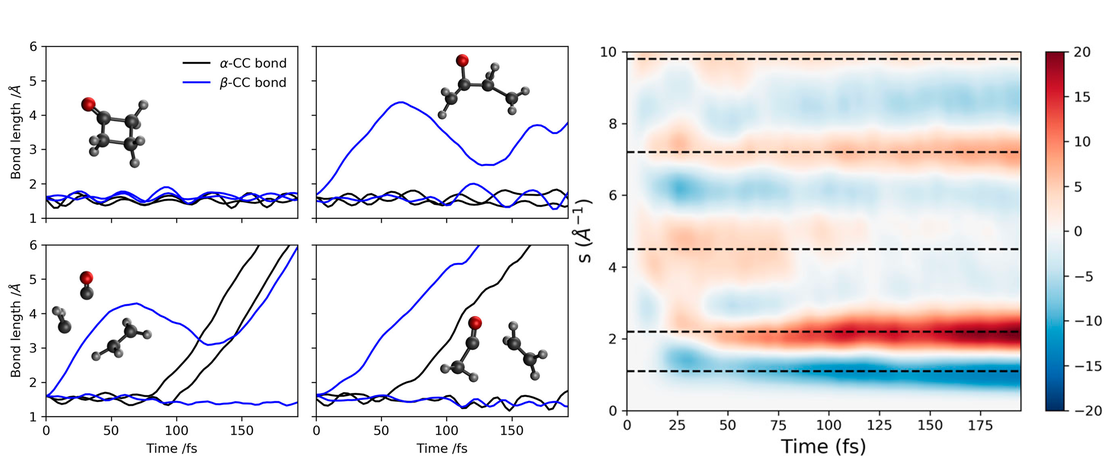
Ultrafast electron diffraction of photoexcited gas-phase cyclobutanone predicted by ab initio multiple cloning simulations
- Dmitry V. Makhov, Lewis Hutton, Adam Kirrander and Dmitry Shalashilin
- Publication , Prediction challenge , Project collaboration
We present the result of our calculations of ultrafast electron diffraction (UED) for cyclobutanone excited into the S2 electronic state, which is based on the non-adiabatic dynamics simulations with the Ab Initio Multiple Cloning (AIMC) method with the electronic structure calculated at the SA(3)-CASSCF(12,12)/aug-cc-pVDZ level of theory…
Read More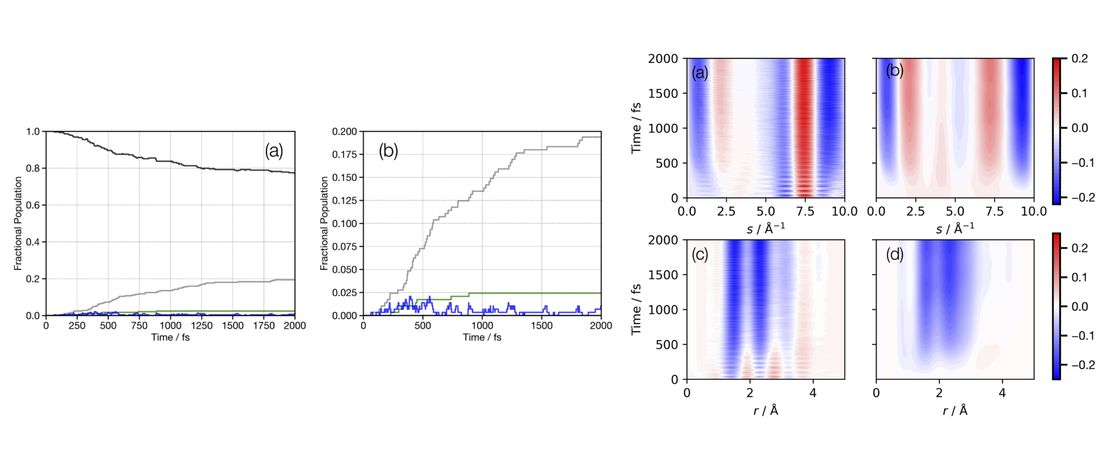
The Photochemistry of Rydberg-Excited Cyclobutanone: Photoinduced Processes and Ground State Dynamics
Owing to ring-strain, cyclic ketones exhibit complex excited-state dynamics with multiple competing photochemical channels active on the ultrafast timescale. While the excitedstate dynamics of cyclobutanone after π ∗ ← n excitation into the lowest-energy excited singlet state (S1) has been extensively studied, the dynamics following 3s ← n excitation into the higher-lying singlet Rydberg (S2) state are less well understood…
Read More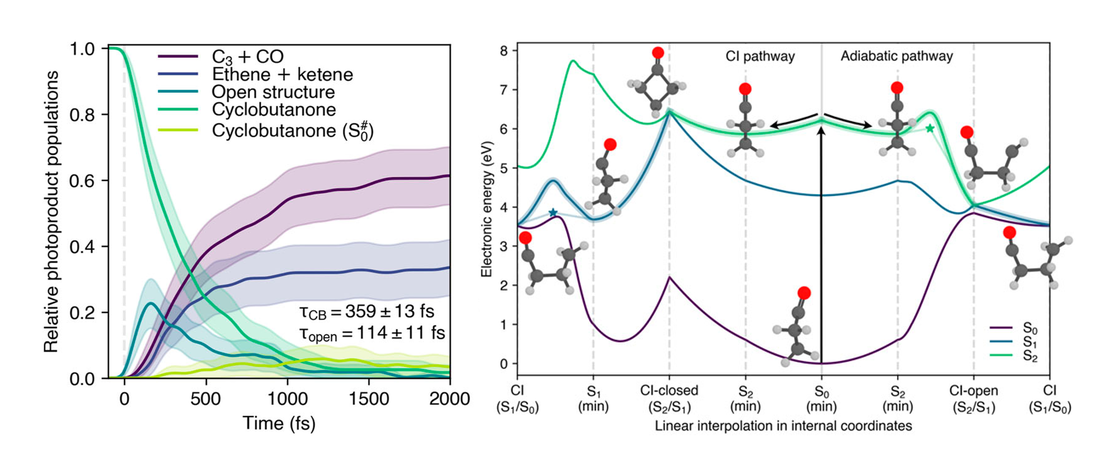
Predicting the photodynamics of cyclobutanone triggered by a laser pulse at 200 nm and its MeV-UED signals—A trajectory surface hopping and XMS-CASPT2 perspective
- Jiří Janoš, Joao Pedro Figueira Nunes, Daniel Hollas, Petr Slavíček and Basile F. E. Curchod
- Publication , Prediction challenge
This work is part of a prediction challenge that invited theoretical/computational chemists to predict the photochemistry of cyclobutanone in the gas phase, excited at 200 nm by a laser pulse, and the expected signal that will be recorded during a time-resolved megaelectronvolt ultrafast electron diffraction (MeV-UED). We present here our theoretical predictions…
Read More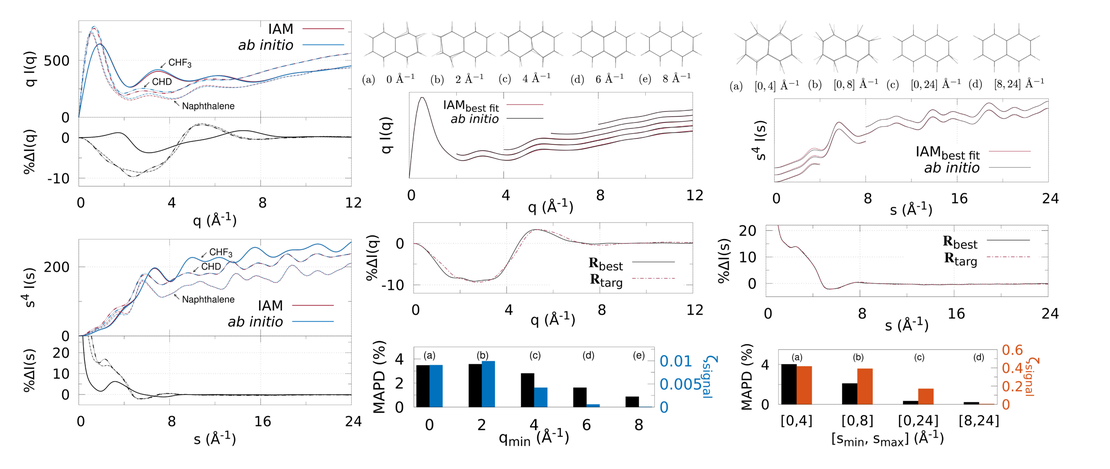
Extracting the electronic structure signal from X-ray and electron scattering in the gas phase
X-ray and electron scattering from free gas-phase molecules is examined using the independent atom model (IAM) and ab initio electronic structure calculations. The IAM describes the effect of the molecular geometry on the scattering, but does not account for the redistribution of valence electrons due to, for instance, chemical bonding…
Read More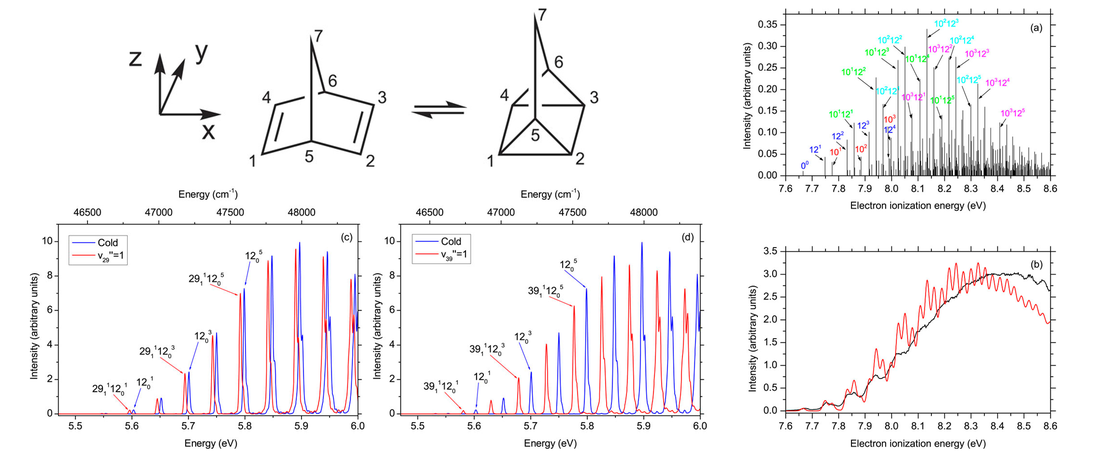
Valence shell electronically excited states of norbornadiene and quadricyclane
- Joseph C. Cooper, David M. P. Holland, Rebecca A. Ingle... Adam Kirrander and Daniel Rolles
- Publication
The absolute photoabsorption cross sections of norbornadiene (NBD) and quadricyclane (QC), two isomers with chemical formula C7H8 that are attracting much interest for solar energy storage applications, have been measured from threshold up to 10.8 eV using the Fourier transform spectrometer at the SOLEIL synchrotron radiation facility…
Read More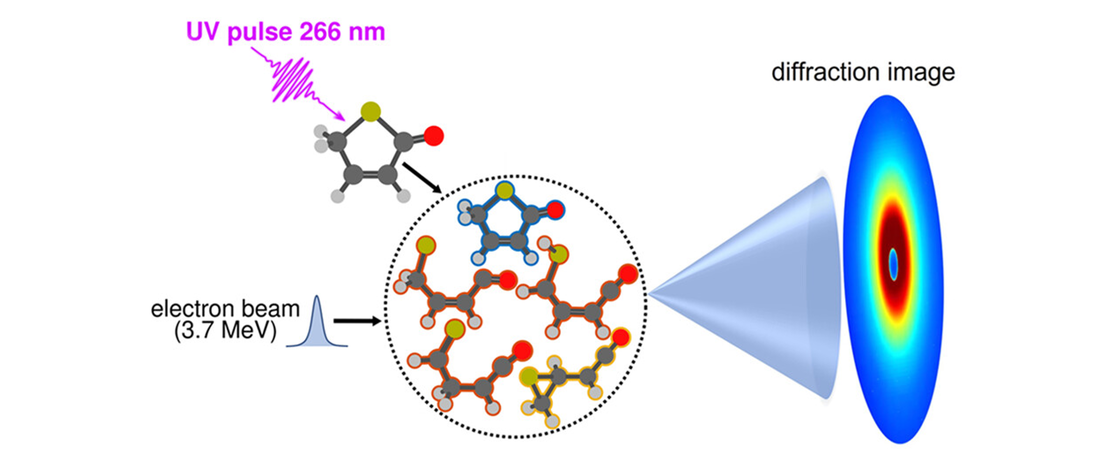
Monitoring the Evolution of Relative Product Populations at Early Times during a Photochemical Reaction
Identifying multiple rival reaction products and transient species formed during ultrafast photochemical reactions and determining their time-evolving relative populations are key steps toward understanding and predicting photochemical outcomes. Yet, most contemporary ultrafast studies struggle with clearly identifying and quantifying competing molecular structures/species among the emerging reaction products…
Read More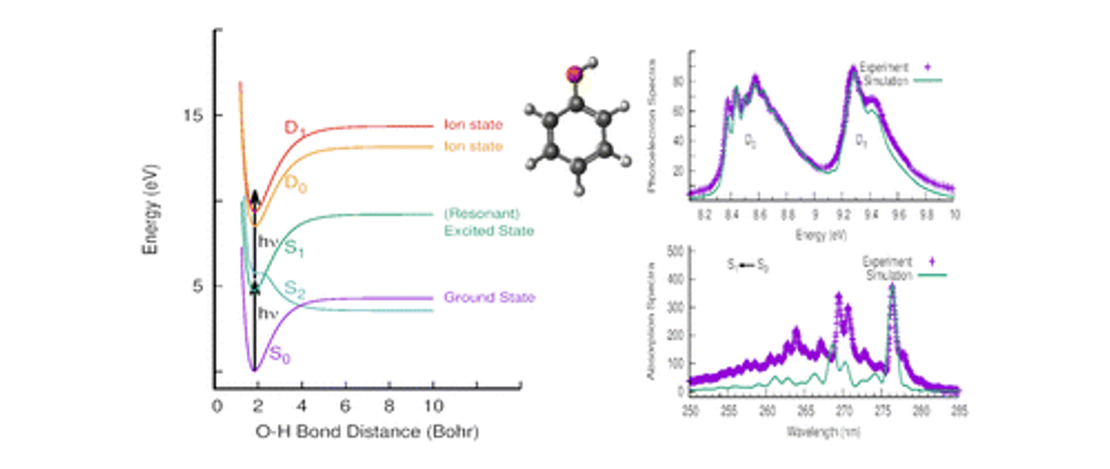
On the multiphoton ionisation photoelectron spectra of phenol
- Diptesh Dey, Joanne L. Woodhouse, Marcus P. Taylor, Helen H. Fielding and Graham A. Worth
- Publication
The phenol molecule is a prototype for non-adiabatic dynamics and the excited-state photochemistry of biomolecules. In this article, we report a joint theoretical and experimental investigation on the resonance enhanced multiphoton ionisation photoelectron (REMPI) spectra of the two lowest ionisation bands of phenol…
Read More