Prediction Through Quantum Dynamics Simulations: Photo-excited Cyclobutanone
- Olivia Bennett, Antonia Freibert, K. Eryn Spinlove and Graham A. Worth
- Prediction challenge , Publication
- May 1, 2024
Abstract:
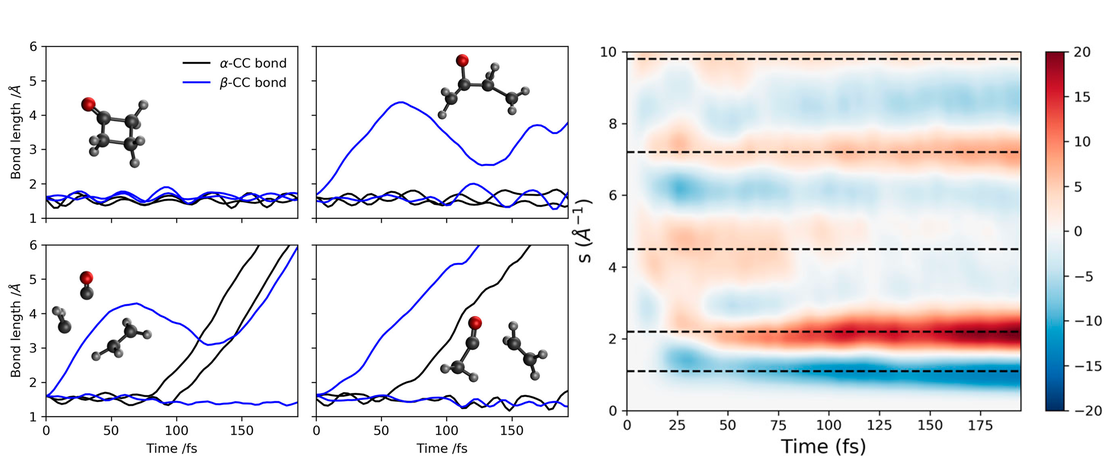
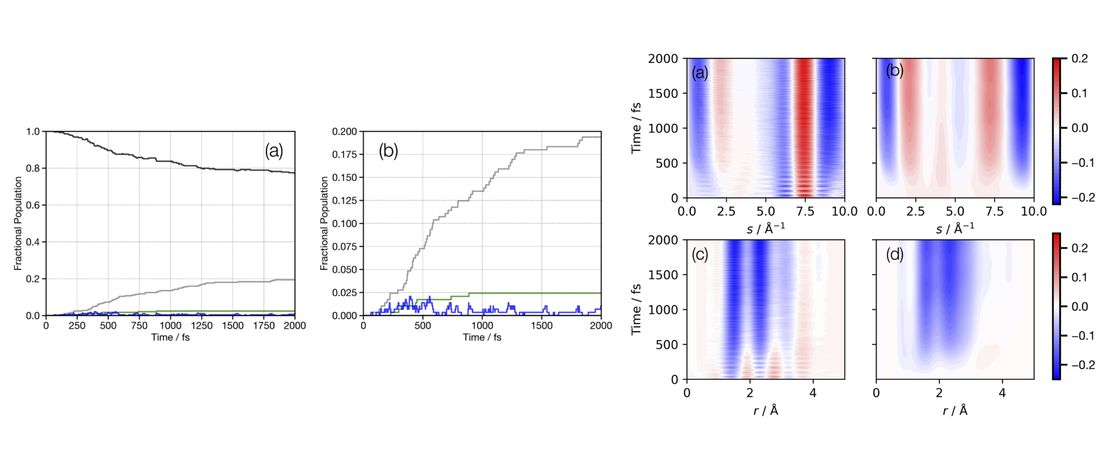
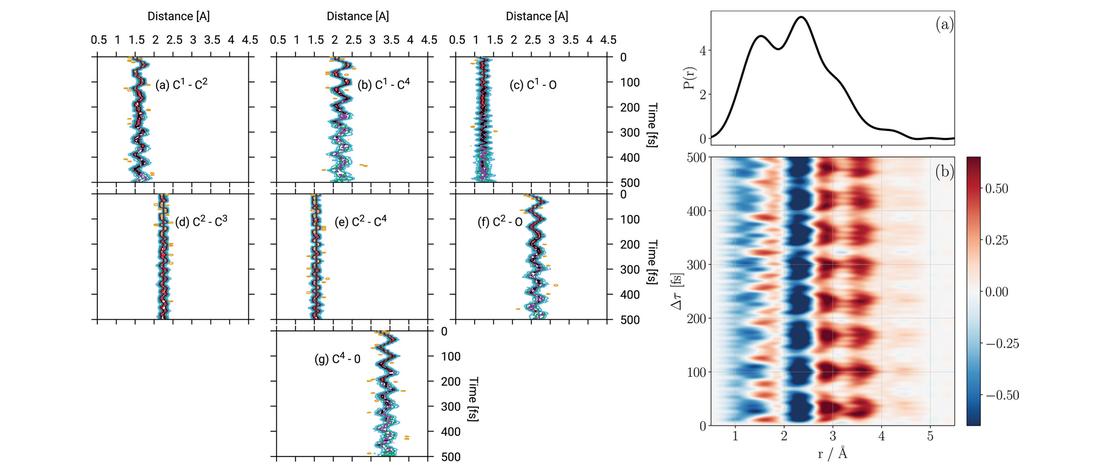
Quantum dynamics simulations are becoming a standard tool for simulating photo- excited molecular systems involving a manifold of coupled states, known as non- adiabatic dynamics. While these simulations have had many successes in explaining experiments and giving details of non-adiabatic transitions, the question remains as to their predictive power. In this work, we present a set of quantum dynam- ics simulations on cyclobutanone, using both grid-based multi-configuration time- dependent Hartree (MCTDH) and direct dynamics variational multi-configuration Gaussian (DD-vMCG) methods. The former used a parameterised vibronic coupling model Hamiltonian and the latter generated the potential energy surfaces on-the-fly. The results give a picture of the non-adiabatic behaviour of this molecule and were used to calculate the signal from a gas-phase ultrafast electron diffraction (GUED) experiment. Corresponding experimental results will be obtained and presented at a later stage for comparison to test the predictive power of the methods. The results show that over the first 500 fs after photo-excitation to the S2 state, cyclobutanone relaxes quickly to the S1 state, but only a small population relaxes further to the S0 state. No significant transfer of population to the triplet manifold is found. It is predicted that the GUED experiments over this time scale will see s signal related mostly to the C-O stretch motion and elongation of the molecular ring along the C-C-O axis.
Additional Resources
DOI:
arXiv:2402.09933, 10.1063/5.0203654
Quick Ref:
J. Chem. Phys., 2024, 160, 17, 174305