Exploring the Influence of Approximations for Simulating Valence Excited X‑ray Spectra
- Thomas J Penfold and Basile F E Curchod
- Publication , Project collaboration
- December 4, 2024
Abstract:
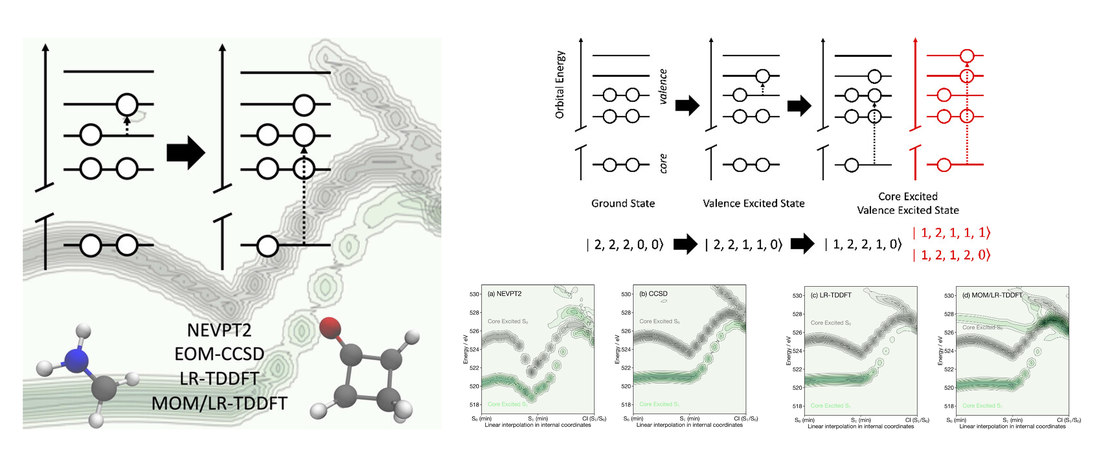
First-principles simulations of excited-state X-ray spectra are becoming increasingly important to interpret the wealth of electronic and geometric information contained within femtosecond X-ray absorption spectra recorded at X-ray Free Electron Lasers (X-FELs). However, because the transition dipole matrix elements must be calculated between two excited states (i.e., the valence excited state and the final core excited state arising from the initial valence excited state) of very different energies, this can be challenging and time-consuming to compute. Herein using two molecules, protonated formaldimine and cyclobutanone, we assess the ability of n-electron valence-state perturbation theory (NEVPT2), equation-of-motion coupled-cluster theory (EOM- CCSD), linear-response time-dependent density functional theory (LR-TDDFT) and the maximum overlap method (MOM) to describe excited state X-ray spectra. Our study focuses in particular on the behavior of these methods away from the Franck−Condon geometry and in the vicinity of important topological features of excited-state potential energy surfaces, namely, conical intersections. We demonstrate that the primary feature of excited-state X-ray spectra is associated with the core electron filling the hole created by the initial valence excitation, a process that all of the methods can capture. Higher energy states are generally weaker, but importantly much more sensitive to the nature of the reference electronic wave function. As molecular structures evolve away from the Franck−Condon geometry, changes in the spectral shape closely follow the underlying valence excitation, highlighting the importance of accurately describing the initial valence excitation to simulate the excited-state X-ray absorption spectra.
Additional Resources
DOI:
Quick Ref:
J. Phys. Chem. A, 2024, 128, 50, 10826