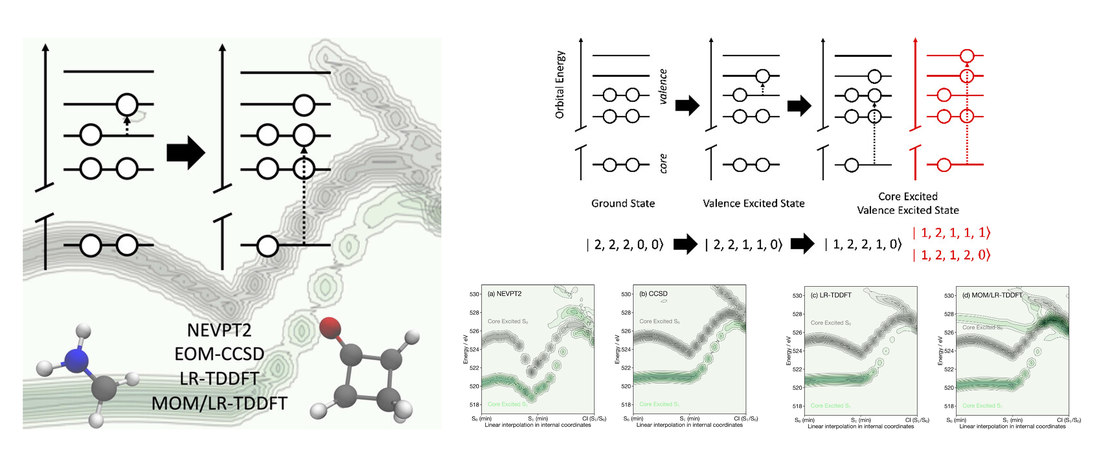
Exploring the Influence of Approximations for Simulating Valence Excited X‑ray Spectra
First-principles simulations of excited-state X-ray spectra are becoming increasingly important to interpret the wealth of electronic and geometric information contained within femtosecond X-ray absorption spectra recorded at X-ray Free Electron Lasers (X-FELs). However, because the transition dipole matrix elements must be calculated between two excited states (i.e., the valence excited state and the final core excited state arising from the initial valence excited state) of very different energies, this can be challenging and time-consuming to compute…
Read More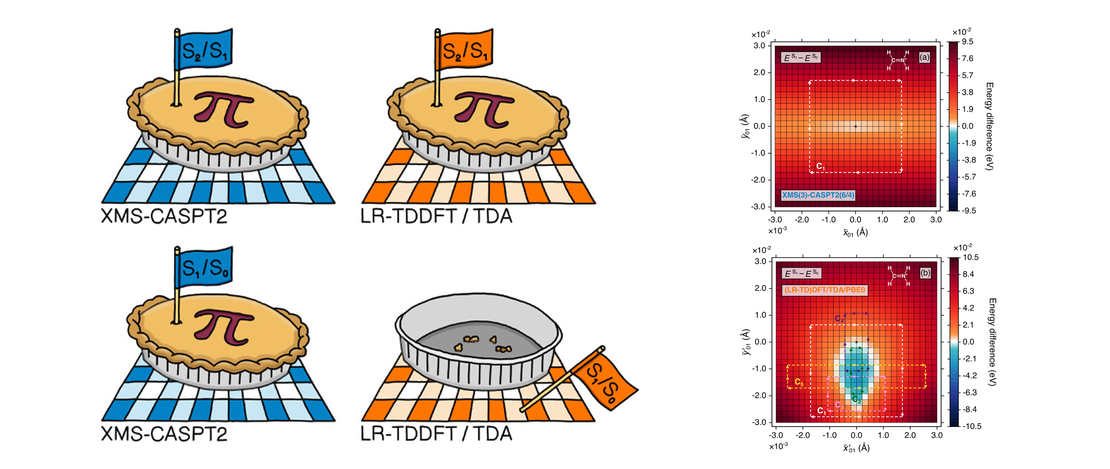
On the Topological Phase around Conical Intersections with Tamm–Dancoff Linear-Response Time-Dependent Density Functional Theory
Regions of nuclear-configuration space away from the Franck–Condon geometry can prove problematic for some electronic structure methods, given the propensity of such regions to possess conical intersections, i.e., (highly connected) points of degeneracy between potential energy surfaces. With the likelihood (perhaps even inevitability) for nonadiabatic dynamics simulations to explore molecular geometries in close proximity to conical intersections, it is vital that the performance of electronic structure methods is routinely examined in this context…
Read More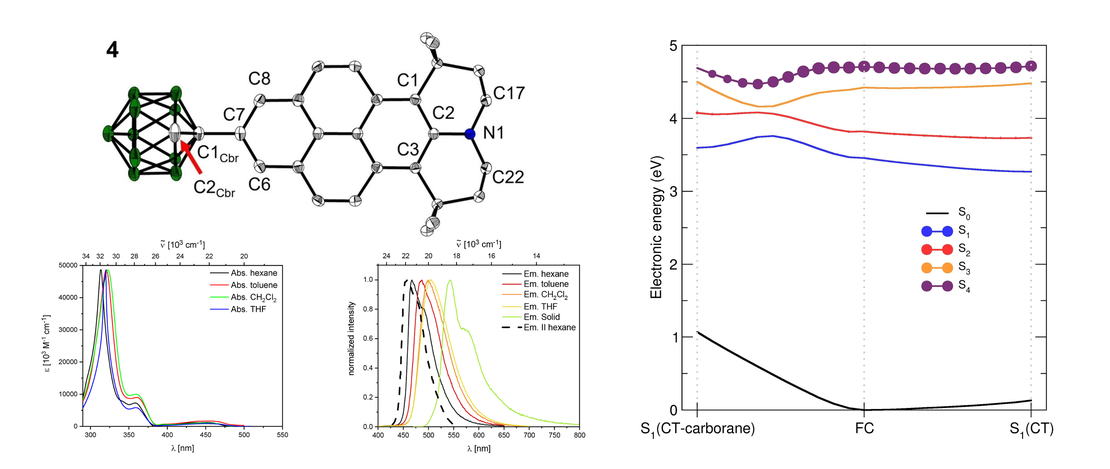
Synthesis, Photophysical and Electronic Properties of a D-π-A Julolidine-Like Pyrenyl-o-Carborane
- Johannes Krebs, Lisa Brändler, Ivo Krummenacher, Alexandra Friedrich, Holger Braunschweig, Maik Finze, Basile F. E. Curchod and Todd B. Marder
- Publication
We synthesized 2-(1-1,2-dicarbadodecaboranyl(12))-6,6,12,12-tetramethyl-7,8,11,12-tetrahydro-6H,10H-phenaleno[1,9-fg]pyrido[3,2,1-ij]quinoline (4), a julolidine-like pyrenyl-o-carborane, with pyrene substituted at the 2,7-positions on the HOMO/LUMO nodal plane. Using solid state molecular structures, photophysical data, cyclic voltammetry, DFT and LR-TDDFT calculations, we compare o-carborane and B(Mes)2 (Mes=2,4,6-Me3C6H2) as acceptor groups…
Read More